X-ray Diffraction (XRD)
X-ray diffraction (XRD) is a powerful analytical technique used to investigate the crystalline structure of materials. XRD provides essential information about the atomic structure, phase composition, and other material properties. XRD is widely employed in fields such as materials science, geology, chemistry, and biology.
Principles of X-ray Diffraction
X-ray diffraction occurs when X-rays interact with the periodic array of atoms in a crystalline material. The periodic arrangement leads to constructive interference at specific angles, resulting in a diffraction pattern. The fundamental principle governing XRD is Bragg’s law, which is mathematically expressed as:
\[ n\lambda = 2d \sin \theta \]
where:
- n is the diffraction order (an integer),
- λ is the wavelength of the X-rays,
- d is the spacing between atomic planes in the crystal,
- θ is half the scattering angle 2θ.
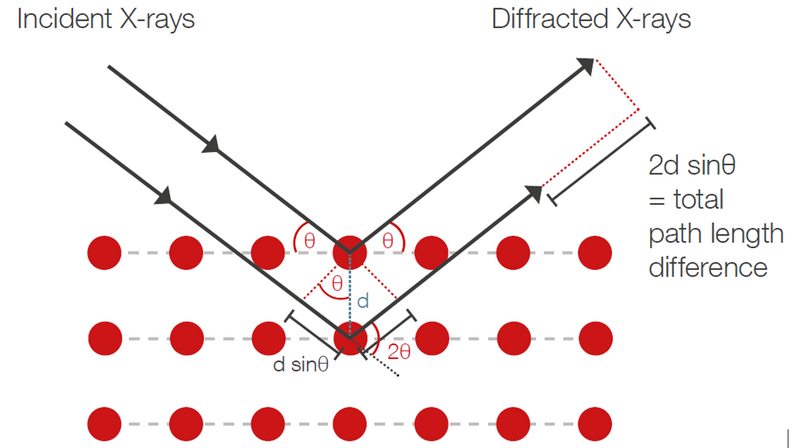
Bragg’s law describes the condition for constructive interference, which occurs when the path length difference between scattered X-rays is an integer multiple of the X-ray wavelength (Figure 1). If this is the case, a diffraction peak is observed at this specific scattering angle 2θ. By varying the scattering angle the so-called diffraction pattern is recorded. Analyzing it allows the determination of the material's crystal structure by measuring the spacing between lattice planes.
X-ray Diffraction Techniques
X-ray diffraction can be performed using different techniques depending on the material being studied and the information required. The most common XRD techniques include:
Powder X-ray diffraction (PXRD)
Powder X-ray diffraction is the most frequently used XRD technique and is ideal for polycrystalline or powdered samples. Since the sample contains many randomly oriented crystallites, X-rays are diffracted in various directions, producing a pattern that can be analyzed for phase identification, quantification, and lattice parameter determination.
Single-crystal X-ray diffraction (SCXRD)
Single-crystal XRD is used to determine the detailed atomic structure of a material by analyzing how X-rays are diffracted by a single crystal. This technique is especially important in crystallography for studying the three-dimensional arrangement of atoms in molecules, such as organic compounds and biological macromolecules like proteins.
High-resolution X-ray diffraction (HRXRD)
High-resolution XRD is used to study materials with very fine structural details, such as thin films and epitaxial layers. HRXRD provides information on strain, lattice mismatch, and defects, which is critical for understanding semiconductor materials and nanostructures.
Grazing-incidence X-ray diffraction (GIXRD)
GIXRD is used to study thin films and surfaces by directing the X-ray beam at a shallow angle to the sample. This technique is useful for analyzing coatings, surface layers, and nanomaterials, where the surface structure may differ from the bulk material.
The Diffractogram and Information obtained from PXRD
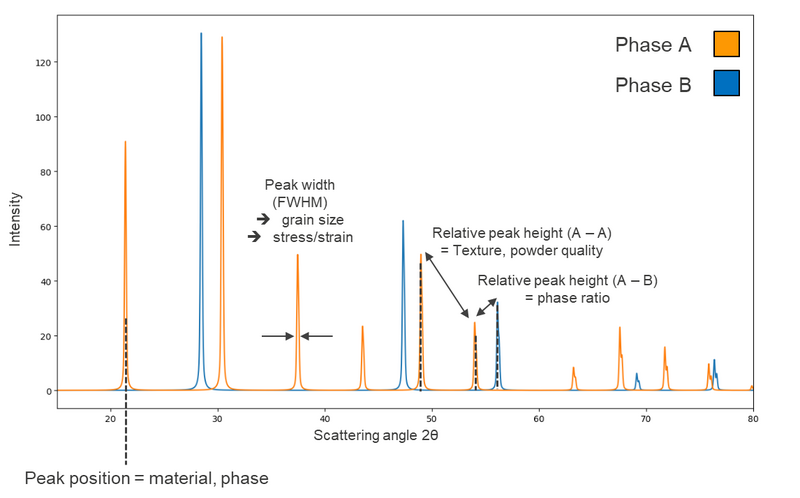
The result of a PXRD experiment is typically presented as a diffractogram – a plot of X-ray intensity (y-axis) versus the diffraction angle (2θ, x-axis). The analysis of a diffractogram yields several key insights:
- Qualitative analysis: Each crystalline material produces a unique diffraction pattern, serving as a "fingerprint" for phase identification. Comparison with databases allows researchers to identify the phase.
- Quantitative analysis: For materials containing multiple phases, XRD is used to quantify the relative amounts of each phase by analyzing the peak intensities/areas.
- Unit cell lattice parameters: XRD characterizes the dimensions of a material's unit cell, the smallest repeating structure in a crystal. This is essential for studying crystallographic properties, especially under non-ambient conditions (e.g., varying temperature or pressure). Also, residual stress has influence on the lattice parameters.
- Crystallite size and strain: The size of crystallites in a sample influences the broadening of diffraction peaks. Small crystallites (less than ~120 nm) produce broader peaks, allowing researchers to estimate crystallite size. XRD can also quantify microstrain in the material.
A schematic representation of a diffractogram and the information contained therein is given in Figure 2.
Additional sophisticated methods of X-ray diffraction exist, which allow to e.g., study residual stress or texture in samples, allowing to gain an even deeper understanding of the samples and their properties. Covering these would exceed the purpose of this page.
Applications of X-ray Diffraction
X-ray diffraction is a versatile technique used across many industries and scientific fields. Its applications range from fundamental research to quality control in manufacturing processes:
- Materials science: XRD is commonly used to characterize the crystalline phases of metals, ceramics, polymers, and composites. It helps in determining the crystallinity, phase composition, and texture of materials, which are critical for their performance in applications such as construction, aerospace, and electronics.
- Geology and mineralogy: XRD is essential in studying minerals, rocks, and soil samples. It is used to identify the mineral phases present in geological samples and to understand the processes that lead to the formation of various minerals under different environmental conditions.
- Chemistry: In chemistry, XRD is used to determine the structure of crystalline compounds, monitor chemical reactions in solid phases, and study catalysts. It is particularly valuable for identifying and quantifying different polymorphs of a compound.
- Pharmaceuticals: XRD plays a crucial role in the pharmaceutical industry, especially in identifying different polymorphic forms of active pharmaceutical ingredients (APIs). The crystal structure of drugs can affect their solubility, stability, and bioavailability, making XRD an important tool in drug development and manufacturing.
- Environmental science: XRD is used in environmental research to study soil and sediment samples. It helps in understanding the distribution of minerals and pollutants, as well as the chemical reactions occurring in natural environments.
- Archaeology and art conservation: XRD is employed in the analysis of ancient artifacts, ceramics, and historical materials. It helps in identifying the composition and origin of materials, aiding in conservation efforts and understanding historical manufacturing techniques.
- Thin films and coatings: In the semiconductor and thin-film industries, XRD is used to analyze the crystalline quality of films, layers, and coatings. GIXRD is particularly useful for studying surface layers and thin films used in microelectronics and photovoltaic devices.
Advantages of X-ray Diffraction
- Non-destructive: XRD is a non-destructive technique, meaning that the sample remains intact after analysis.
- Wide applicability: XRD can be used to study a wide range of materials, from metals and ceramics to polymers, minerals, and biological macromolecules.
- High precision: XRD provides precise information about atomic arrangements and crystal structures.
- Rapid and efficient: The analysis can be performed relatively quickly, making XRD suitable for both research and industrial quality control.
Limitations of X-ray Diffraction
- Crystallinity requirement: XRD is effective only for crystalline materials. Amorphous materials, which lack long-range order, do not produce clear diffraction patterns.
- Sample size, amount and quality: For single-crystal XRD, high-quality crystals of sufficient size are required, which may not always be possible for certain materials. In powder XRD, a homogenous crystallite size distribution, random orientation distribution, and a certain crystallite size are necessary to obtain high-quality data. Sufficient material has to be available to collect diffractograms in a reasonable time.
- Phase overlap: In complex mixtures, overlapping diffraction peaks can make it difficult to distinguish between different phases in powder XRD.
Conclusion
X-ray diffraction is an essential tool for understanding the structure and properties of crystalline materials. From phase identification to the determination of crystallite size, lattice strain, and crystal structure, XRD provides detailed insights into materials that are crucial for various applications in science and industry. Its non-destructive nature, versatility, and high precision make it indispensable in fields ranging from materials science to pharmaceuticals and environmental research.

The Non-ambient Guide
This free guide gives you a general introduction to non-ambient X-ray diffraction (NA-XRD). The content spans from instrumentation, analysis, and data interpretation to applications. The document is not explicitly dedicated to one specific non-ambient XRD instrument or one particular application area, but aims to give a global overview of the main instrumentation and applications.
References
- Touloukian, Y.S., (1977). Thermophysical Properties of Matter, Vol 13, Thermal Expansion of Nonmetallic Solids
- Bragg, W. L., & Bragg, W. H. (1913). The Reflection of X-rays by Crystals. Proceedings of the Royal Society A, 88(605), 428-438.
- Cullity, B. D., & Stock, S. R. (2001). Elements of X-ray Diffraction. Pearson Education.
- Klug, H. P., & Alexander, L. E. (1974). X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials. Wiley.